
Complexity Thoughts: Issue #83
Unraveling complexity: building knowledge, one paper at a time
If you find value in #ComplexityThoughts, consider helping it grow by subscribing and sharing it with friends, colleagues or on social media. Your support makes a real difference.
→ Don’t miss the podcast version of this post: click on “Spotify/Apple Podcast” above!
From the Lab
Unraveling the emergence of collective behavior in networks of cognitive agents
Three years ago I was wondering whether LLM could act in “social groups” of any sort, and how such networks would resemble real ones.
A few months later, a brilliant student, Nicola Zomer, who was following my course on the physics of complex networks, came to me to ask for a thesis on exactly this question. Now, that work is published in NPJ Artificial Intelligence, and my warmest congratulations go to Nicola.
What makes this study especially timely is that it asks a question that is becoming central for AI: what happens when we stop looking at a single intelligent agent in isolation and start looking at many of them interacting as a collective?
Our work shows that giving agents LLM-based reasoning does not automatically produce better collective intelligence. In some tasks, a single cognitive agent can outperform a classical search process because it can detect patterns and exploit past information more effectively.
But when many such agents interact, the picture becomes much richer: they can also converge too quickly, imitate one another and get trapped in poor collective solutions.
This is why I think that or paper is important. It shows that the behavior of an artificial society is shaped not only by how smart each agent is, but also by how agents communicate, who listens to whom and how information flows through the network. In other words, the architecture of interaction is not a technical detail: it is part of the intelligence of the system.
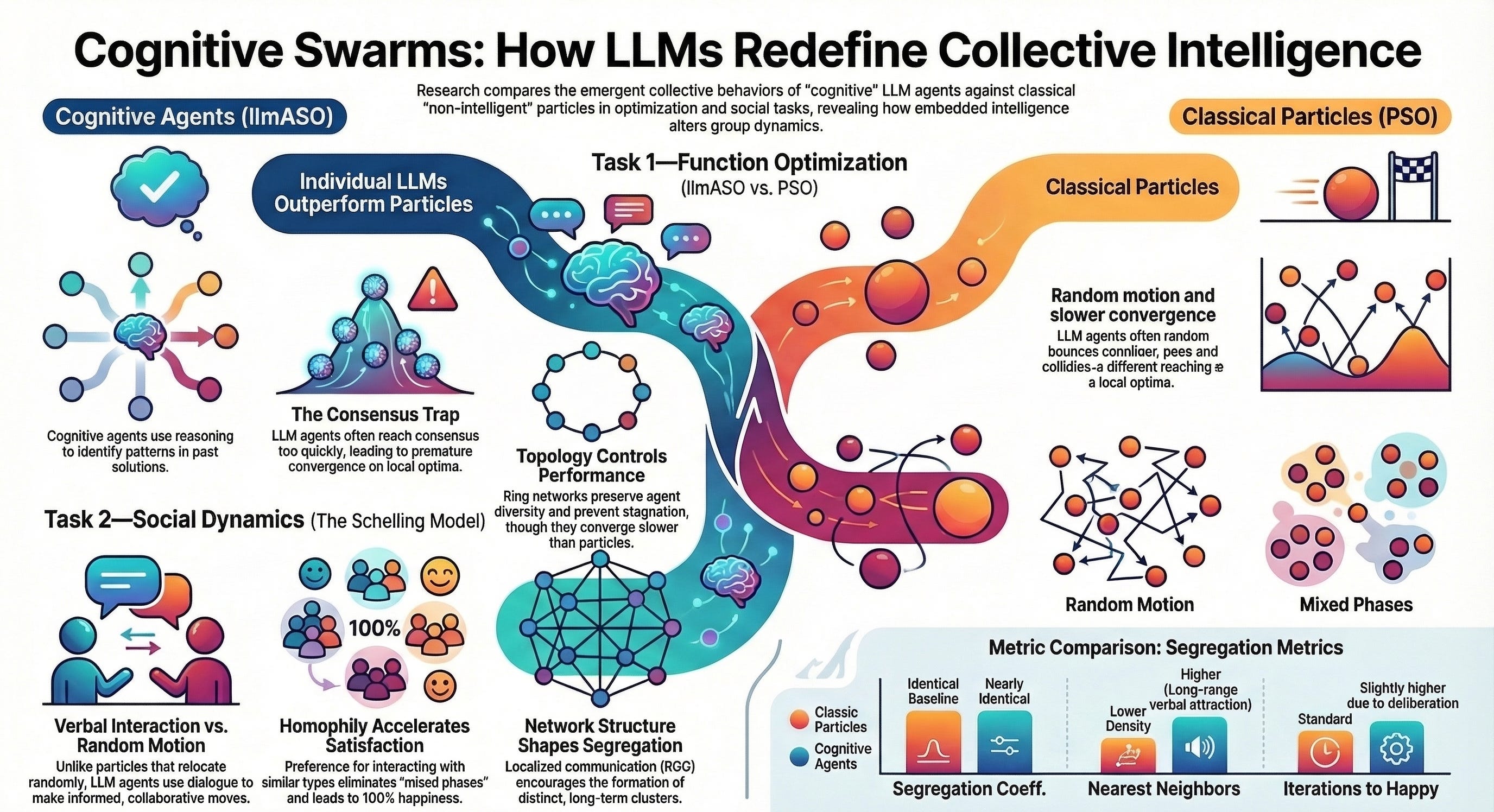
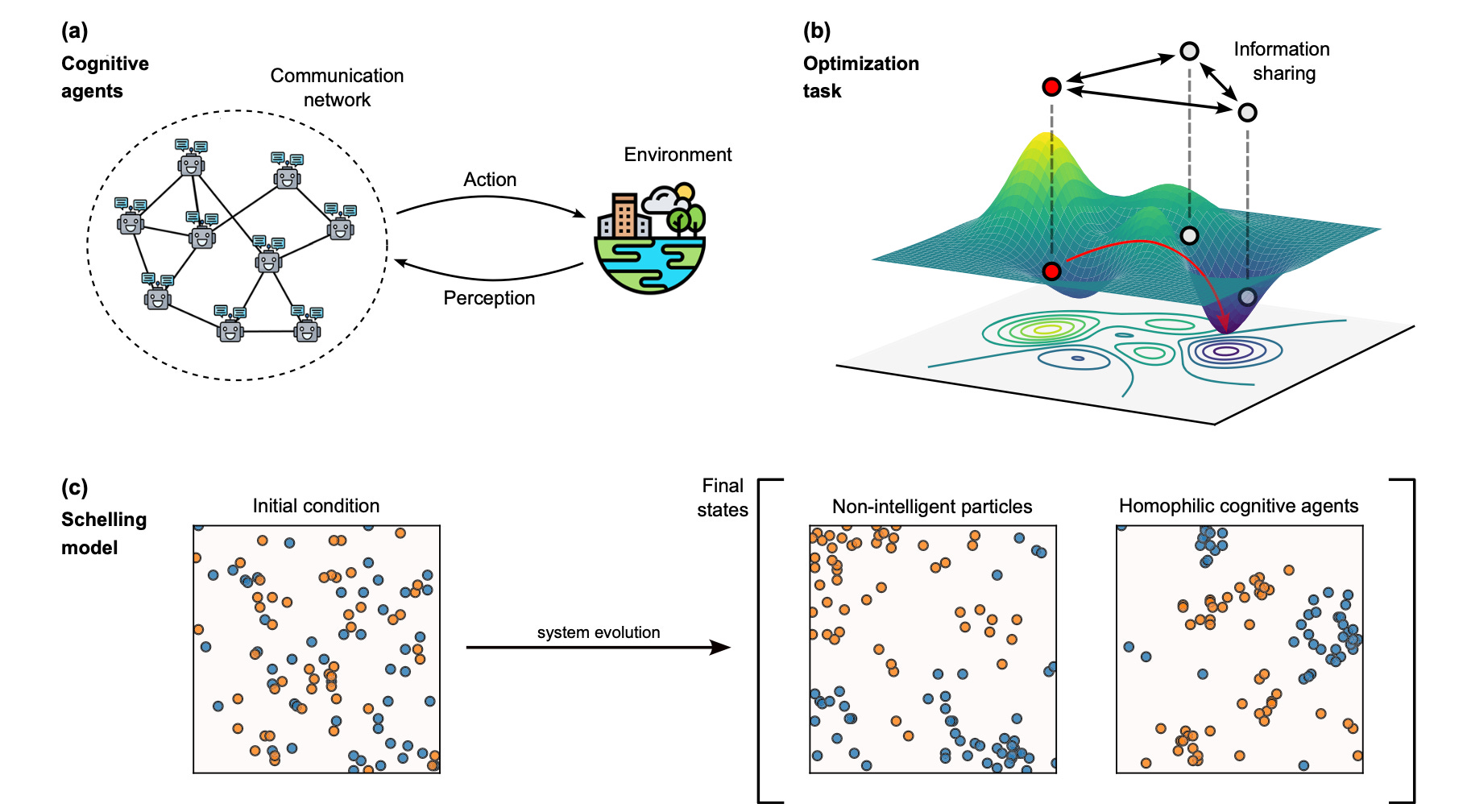
A second key result is that collective behavior depends strongly on network structure. In optimization, collaboration among LLM agents can recover the global solution more reliably than isolated agents, but only when the communication pattern preserves enough diversity. In social self-organization, LLM agents do not simply create new macroscopic behavior by default; instead, meaningful differences emerge when interactions are local and socially selective.
For those working in AI, I think the take-home messages are clear:
intelligence at the individual level does not guarantee intelligence at the collective level;
communication topology is a first-class design variable for multi-agent systems;
understanding emergence, consensus, diversity and coordination will be essential if we want robust agentic AI.
This is exactly where complex systems science can make a difference!
If you are curious, read also this post, where I better put the work in the context of agentic AI:

Cognitive agents, powered by Large Language Models (LLMs), possess advanced reasoning and communication capabilities that fundamentally distinguish them from non-cognitive particles, which rely solely on formal rules. While their ability to replicate human individual and social behaviors is still under scrutiny, the impact of their embedded “intelligence” on emergent behaviors remains poorly understood. Here, by comparing cognitive agents with classic particles, we investigate how LLM capabilities shape emergent phenomena in two tasks: function optimization and social organization emerging from the Schelling model of segregation. To this aim, we introduce LLM Agent Swarm Optimization (llmASO), where a swarm of interacting LLM agents acts as an optimizer. Our findings reveal that, while individual agents outperform particles in decision-making, their consensus tendencies and ability to exploit patterns can make them prone to premature convergence. Adjusting network topology can alleviate this effect, but typically at the cost of slower overall convergence compared to classical Particle Swarm Optimization (PSO). In contrast, in the Schelling model, we demonstrate that local interactions and homophilic mechanisms allow cognitive agents to generate distinct emergent behaviors, underscoring the importance of realistic communication architectures in complex social simulations. This work clarifies how LLM capabilities introduce new mechanisms for collective behavior and has implications for future applications of LLM agents in swarm robotics, social experiments, and complex decision-making tasks.
Artificial and biological systems
Bringing the genetically minimal cell to life on a computer in 4D
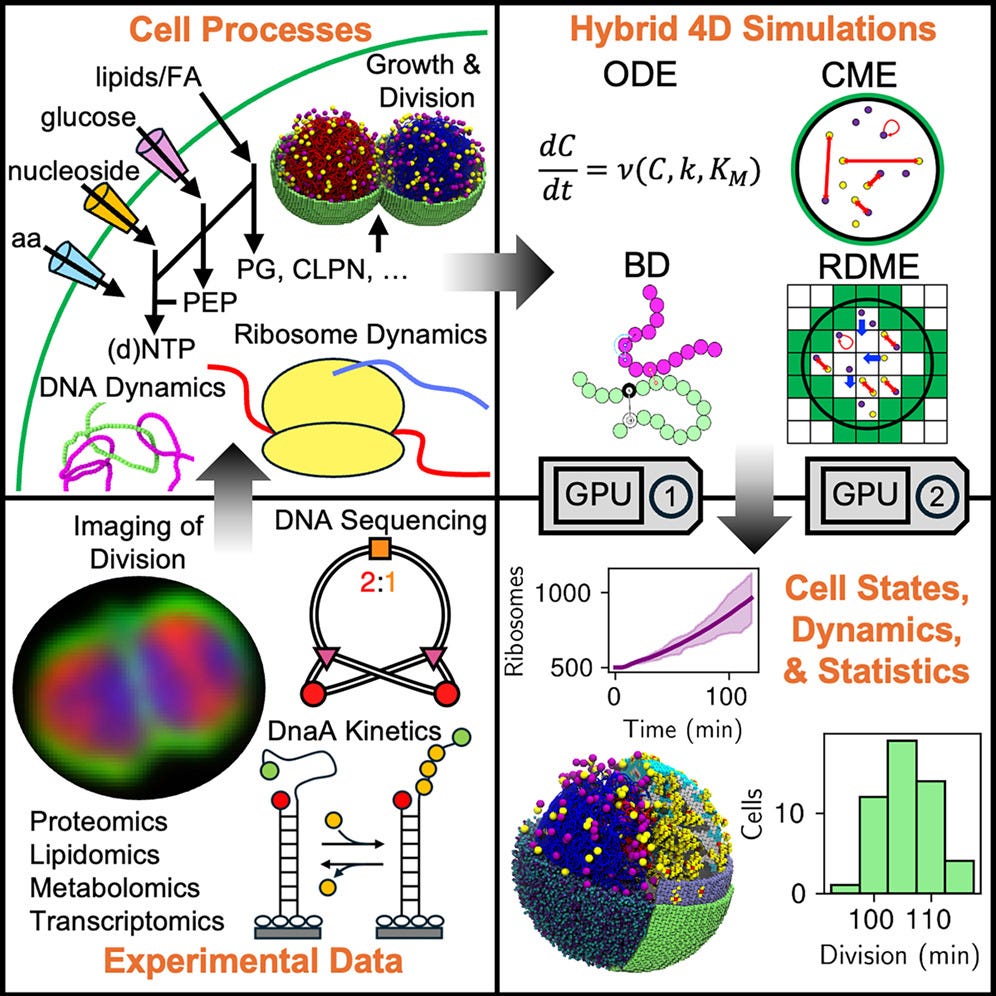
We present a whole-cell spatial and kinetic model for the 100 min cell cycle of the genetically minimal bacterium JCVI-syn3A. We simulate the complete cell cycle in 4D (space and time), including all genetic information processes, metabolic networks, growth, and cell division. By integrating hybrid computational methods, we model the dynamics of morphological transformations. Growth is driven by insertion of lipids and membrane proteins and constrained by fluorescence imaging data. Chromosome replication and segregation are controlled by the essential structural maintenance of chromosome proteins, analogous to condensin (SMC) and topoisomerase proteins in Brownian dynamics simulations, with replication rates responding to deoxyribonucleotide triphosphate (dNTP) pools from metabolism. The model captures the origin-to-terminus ratio measured in our DNA sequencing and recovers other experimental measurements, such as doubling time, mRNA half-lives, protein distributions, and ribosome counts. Because of stochasticity, each replicate cell is unique. We predict not only the average behavior of partitioning to daughter cells but also the heterogeneity among them.
Gene regulatory networks: from correlative models to causal explanations
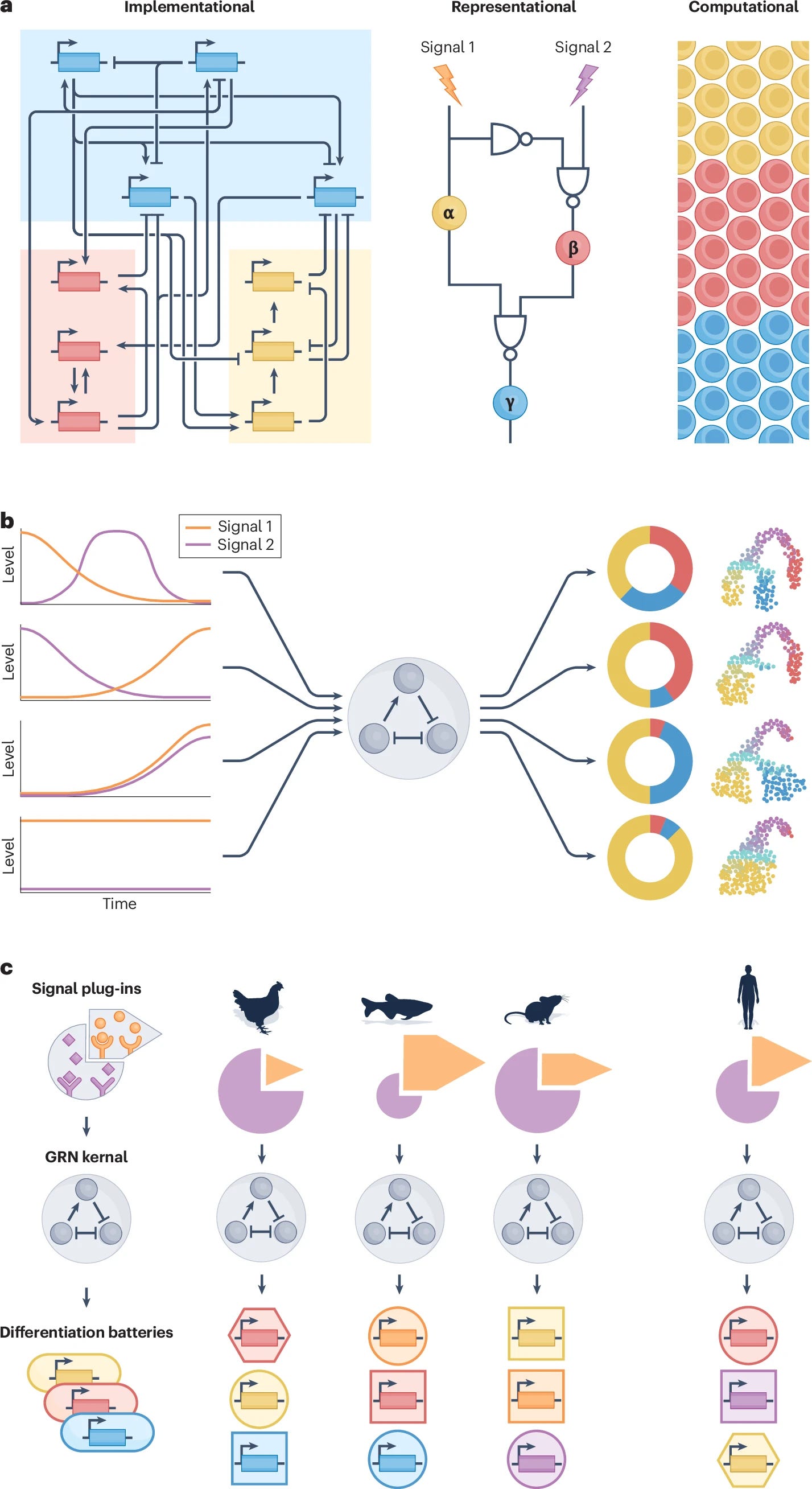
Gene regulatory networks (GRNs) explain how the genome controls cellular behaviour and tissue morphogenesis, serving to connect molecular mechanism to functional output. Single-cell technologies now provide descriptions of these networks with unprecedented detail, but this advance has also revealed gene regulatory systems that are too complex for our existing conceptual frameworks. GRNs, which should provide mechanistic explanations, are increasingly reduced to statistical correlations — ‘hairballs’ that fail to capture molecular causation. Here, we explore why this dilemma exists and propose a path forward. We argue that methods in ‘representation learning’ can be used to model GRNs, without needing to capture every molecular detail. For this framework, we advocate three linked principles: models must be inherently mechanistic, with structures grounded in cellular and evolutionary biology; molecular principles and constraints must be used to reduce the solution space for learning GRN models; and more sophisticated forms of experimental perturbation and synthetic biological engineering are needed to train models and test predictions. By reimagining GRNs through these principles, we can bridge the gap from data abundance to new conceptual understanding.
Genome modelling and design across all domains of life with Evo 2
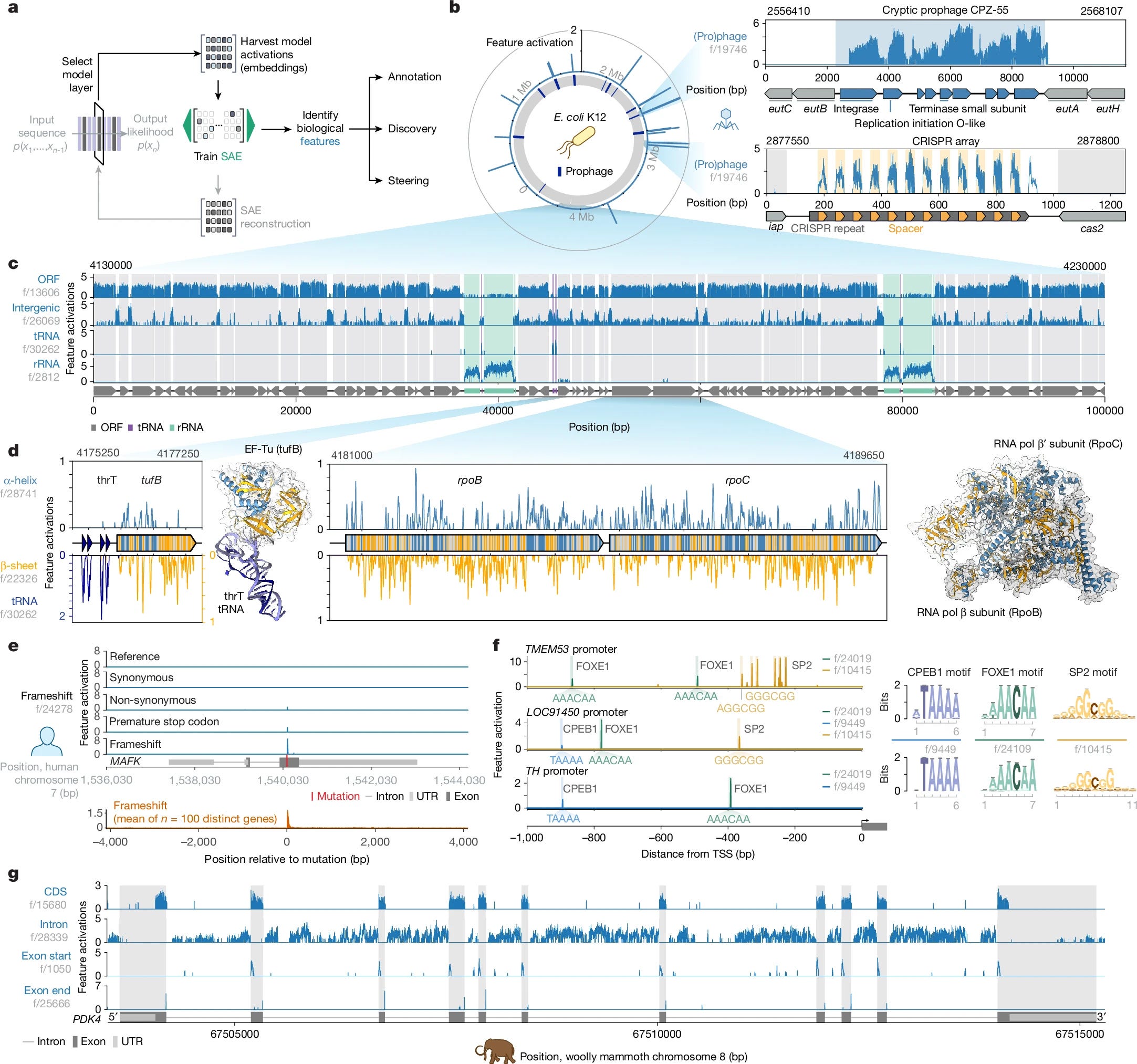
All of life encodes information with DNA. Although tools for genome sequencing, synthesis and editing have transformed biological research, we still lack sufficient understanding of the immense complexity encoded by genomes to predict the effects of many classes of genomic changes or to intelligently compose new biological systems. Artificial intelligence models that learn information from genomic sequences across diverse organisms have increasingly advanced prediction and design capabilities1,2. Here we introduce Evo 2, a biological foundation model trained on 9 trillion DNA base pairs from a highly curated genomic atlas spanning all domains of life to have a 1 million token context window with single-nucleotide resolution. Evo 2 learns to accurately predict the functional impacts of genetic variation—from noncoding pathogenic mutations to clinically significant BRCA1 variants—without task-specific fine-tuning. Mechanistic interpretability analyses reveal that Evo 2 learns representations associated with biological features, including exon–intron boundaries, transcription factor binding sites, protein structural elements and prophage genomic regions. The generative abilities of Evo 2 produce mitochondrial, prokaryotic and eukaryotic sequences at genome scale with greater naturalness and coherence than previous methods. Evo 2 also generates experimentally validated chromatin accessibility patterns when guided by predictive models3,4 and inference-time search. We have made Evo 2 fully open, including model parameters, training code5, inference code and the OpenGenome2 dataset, to accelerate the exploration and design of biological complexity.
Population dynamics
Integrated framework to study genomic surveillance of selective sweeps in multivariants dynamics
Pandemics are often shaped by the competition of multiple pathogen variants, yet existing models rarely connect epidemic dynamics, genomic surveillance, and population immunity within a unified framework. We develop an integrated modeling framework that couples multivariant epidemic dynamics, mechanistic genomic dominance, and probabilistic surveillance. We show that variant dominance follows selective sweeps described by multilogistic equations, with growth rates modulated by population immune histories. Moreover, we show that the variant emergence timing and the dynamics of prior variants influence whether new variants are detected quickly or remain undetected. Our results are validated by fitting epidemiological and genomic data from the spread of SARS-CoV-2 variants across multiple countries, highlighting its relevance for genomic epidemiology and surveillance-informed response.
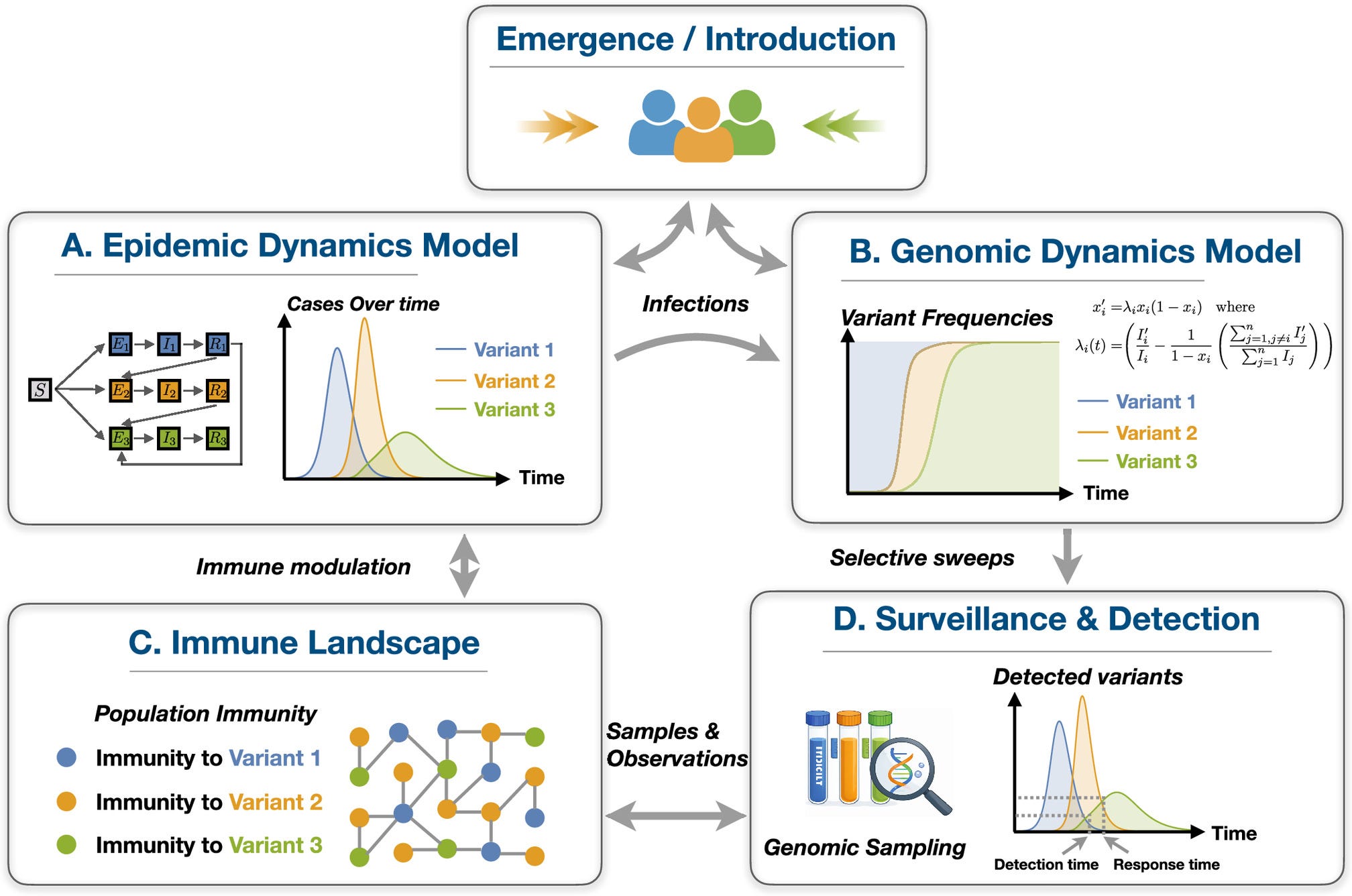
Pandemics often involve complex transmission dynamics in which epidemiological surveillance is essential but not sufficient for containment, as resurgence may be driven by emerging or imported variants. Rapidly evolving pathogens produce complex disease dynamics driven by emerging variants often differing in their transmissibility, immune escape, and cross-infection. These processes influence individuals’ immune life histories, producing highly dynamic immune landscapes that modulate the emergence and dominance of novel variants. We develop an integrated modeling framework that couples multivariant mean-field epidemic modeling with a mechanistic genomic dominance model and a probabilistic surveillance model. This study examines how variant emergence timing, infectiousness advantage, and cross-infection jointly shape epidemic trajectories, immune landscapes, and genomic composition. Our results demonstrate that the dominance dynamics of cocirculating variants correspond to a selective sweep characterized by a system of multilogistic equations driven by population immunity. Moreover, we show that the detection time of newly introduced variants can be accelerated or delayed depending on their emergence conditions and the prevailing variant landscape. Finally, we demonstrate that the effectiveness of response strategies depends critically on the evolving genomic composition of the outbreak, highlighting trade-offs between surveillance sensitivity and intervention timing. We validate our framework by jointly fitting epidemiological and genomic data from the spread of the Ancestral, Alpha, Gamma, and Delta variants in the United States, Denmark, the United Kingdom, and Canada. The results provide a quantitative foundation for linking epidemic dynamics, genomic surveillance, and immune life histories, advancing the development of genomic epidemiology for multivariant outbreaks.
Characterization and forecast of global influenza subtype dynamics
The subtype composition of seasonal influenza waves varies in space and time. Influenza subtypes A/H1N1, A/H3N2 and B tend to have different impacts on population groups; therefore, understanding the drivers of their cocirculation and anticipating their composition is important for epidemic preparedness. FluNet provides data on influenza specimens by subtype for more than 150 countries. However, owing to surveillance variations across countries, global analyses usually focus on subtype compositions, a kind of data difficult to treat with advanced statistical methods. We used compositional data analysis to circumvent the problem and study trajectories of annual subtype compositions of countries. Here we first examine global trends from 2000 to 2023. We identify a few seasons which stood out for the strong within-country subtype dominance due to either a new virus/clade taking over (2003/2004 season, A/H1N1pdm pandemic) or subtypes’ spatial segregation (coronavirus disease 2019 pandemic). Second, we show that geographical factors, most notably international mobility, concurred in shaping countries’ composition trajectories between 2010 and 2019. Trajectories clustered in two macroregions characterized by subtype alternation versus persistent mixing. Finally, we define five algorithms for forecasting the next year’s composition and found that incorporating the global history of subtype composition in a Bayesian hierarchical vector autoregressive model improved predictions compared with naive methods. The joint analysis of spatiotemporal dynamics of influenza subtypes worldwide reveals a hidden structure in subtype circulation that can be used to improve predictions of the subtype composition of next year’s epidemic according to place.
Genotype networks drive oscillating endemicity and epidemic trajectories in viral evolution
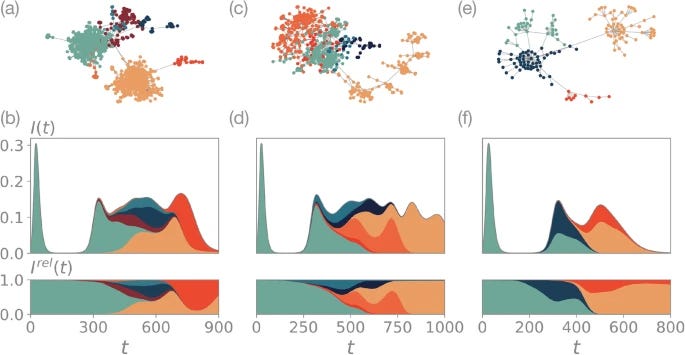
Rapidly evolving viruses use antigenic drift as a key mechanism to evade host immunity and persist in real populations. While traditional models of antigenic drift and epidemic spread rely on low-dimensional antigenic spaces, genomic surveillance data reveal that viral evolution produces complex antigenic genotype networks with hierarchical modular structures. In this study, we present an eco-evolutionary framework in which viral evolution and population immunity dynamics are shaped by the structure of antigenic genotype networks. Using synthetic networks, we demonstrate that network topology alone can drive transitions between stable endemic states and recurrent seasonal epidemics. Furthermore, our results show how the integration of the genotype network of the H3N2 influenza in our model allows for estimating the emergence times of various haplotypes resulting from its evolution. Our findings underscore the critical role of the topology of genotype networks in shaping epidemic behavior and, besides, provide a robust framework for integrating real-world genomic data into predictive epidemic models.
Immunity-induced criticality of the genotype network of influenza A (H3N2) hemagglutinin
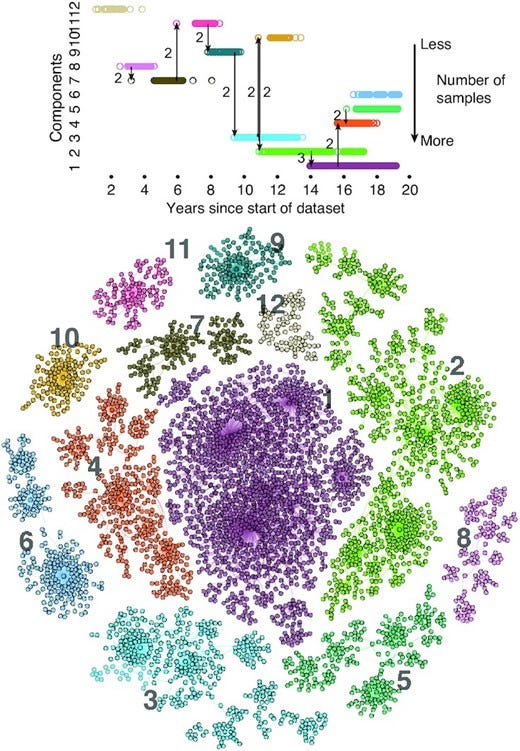
Seasonal influenza kills hundreds of thousands every year, with multiple constantly changing strains in circulation at any given time. A high mutation rate enables the influenza virus to evade recognition by the human immune system, including immunity acquired through past infection and vaccination. Here, we capture the genetic similarity of influenza strains and their evolutionary dynamics with genotype networks. We show that the genotype networks of influenza A (H3N2) hemagglutinin are characterized by heavy-tailed distributions of module sizes and connectivity indicative of critical behavior. We argue that (i) genotype networks are driven by mutation and host immunity to explore a subspace of networks predictable in structure and (ii) genotype networks provide an underlying structure necessary to capture the rich dynamics of multistrain epidemic models. In particular, inclusion of strain-transcending immunity in epidemic models is dependent upon the structure of an underlying genotype network. This interplay is consistent with self-organized criticality where the epidemic dynamics of influenza locates critical regions of its genotype network. We conclude that this interplay between disease dynamics and network structure might be key for future network analysis of pathogen evolution and realistic multistrain epidemic models.
→ Please, remind that if you find value in #ComplexityThoughts, you might consider helping it grow by subscribing, or by sharing it with friends, colleagues or on social media. See also this post to learn more about this space.