
Complexity Thoughts: Issue #85
Unraveling complexity: building knowledge, one paper at a time
If you find value in #ComplexityThoughts, consider helping it grow by subscribing and sharing it with friends, colleagues or on social media. Your support makes a real difference.
→ Don’t miss the podcast version of this post: click on “Spotify/Apple Podcast” above!
Evolution
Epistasis and co-adaptation in bacterial genome evolution
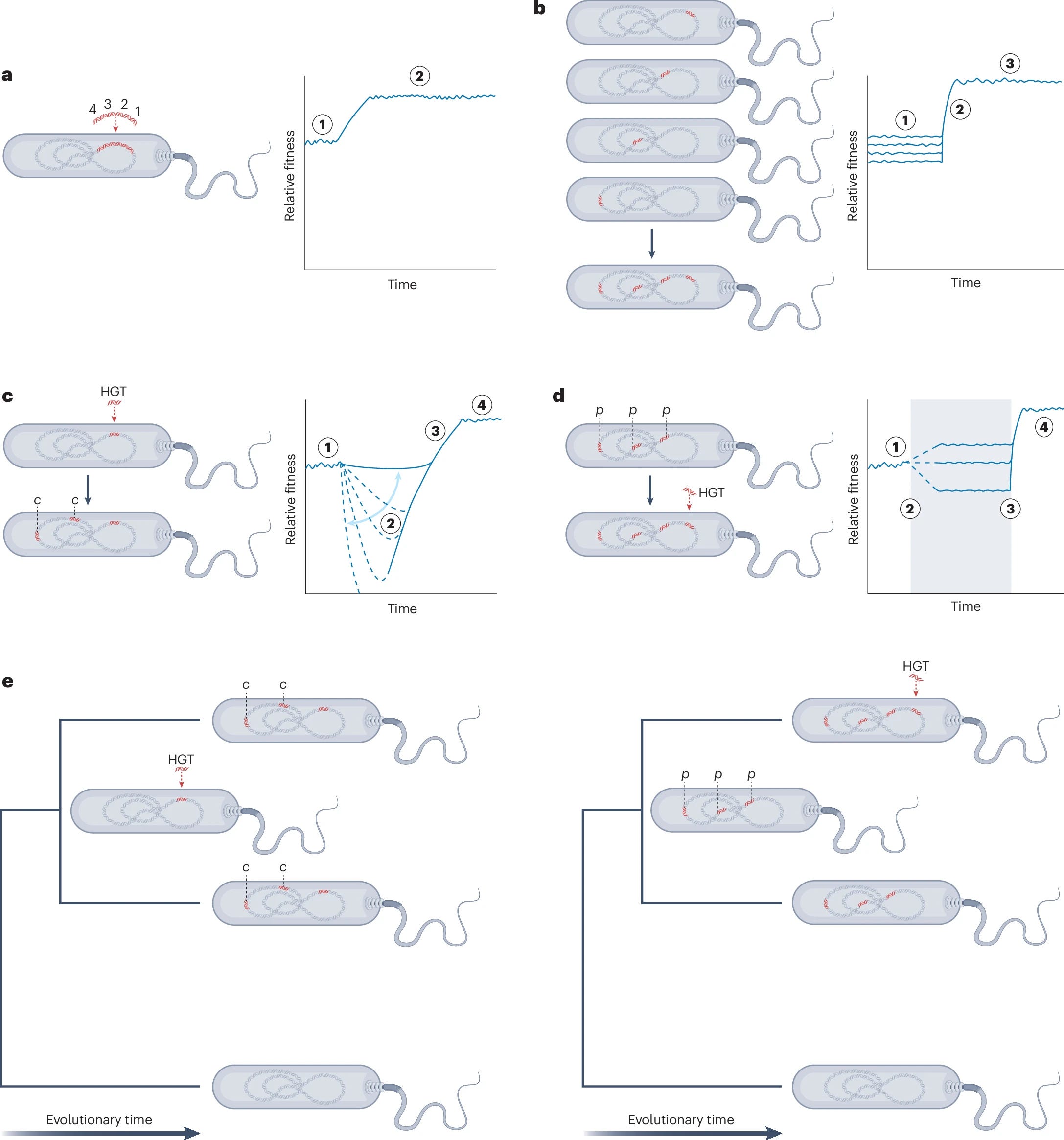
Precise genotype–phenotype mapping is essential in applied microbiology, from engineering genetically modified strains to developing tailored strategies for antimicrobial therapies. Comparative genomics often treats genes as independent contributors to phenotypes, and gene knockout and complementation remain the gold standard to validate genotype–phenotype associations in microorganisms. However, genes do not act in isolation, and complex gene–gene interactions, that is, epistatic interactions, are essential for the evolution and function of bacterial genomes. Recent advances in high-throughput genomics and experimental techniques have enabled systematic screens of epistasis in bacteria at scale, revealing mechanisms underlying epistasis and co-adaptation in laboratory and wild populations. Here we review how microbial genomics is moving beyond gene-centric models towards integrated analyses of potentiating, compensatory and context-dependent variation. The timely incorporation of interaction-based perspectives into population-scale analyses will improve genotype–phenotype mapping and the understanding of the complex traits that shape the microbial world.
Asgard archaea and the hidden complexity of our microbial origins
Long before plants and animals altered Earth’s surface, microbial cells were already testing ways to manage energy, build internal organization and survive changing chemistry. These two spapers use newly recovered genomes from Asgard archaea (we have already covered them some Issues ago), the closest known archaeal relatives of eukaryotes, to refine what that ancestral cell may have been capable of.
One analysis expands the known diversity of Heimdallarchaeia and finds genes linked to oxygen use, detoxification, haem synthesis and respiratory machinery, suggesting that aerobic metabolism or hydrogen-based energy strategies may have shaped the lineage from which eukaryotes emerged.
The other study looks beyond DNA similarity by predicting protein shapes, revealing hundreds of Asgard proteins that resemble eukaryotic components involved in information processing, intracellular transport and compartment formation.
Together, the results suggest that the ancestor shared by Asgard archaea and eukaryotes had a richer molecular toolkit than sequence comparisons alone implied. This does not reconstruct a modern eukaryotic cell, but it narrows the gap between simple prokaryotic life and the cellular complexity that later supported animals, plants, fungi, and protists. You might want to read also the coverage by Phys.org
Oxygen metabolism in descendants of the archaeal-eukaryotic ancestor
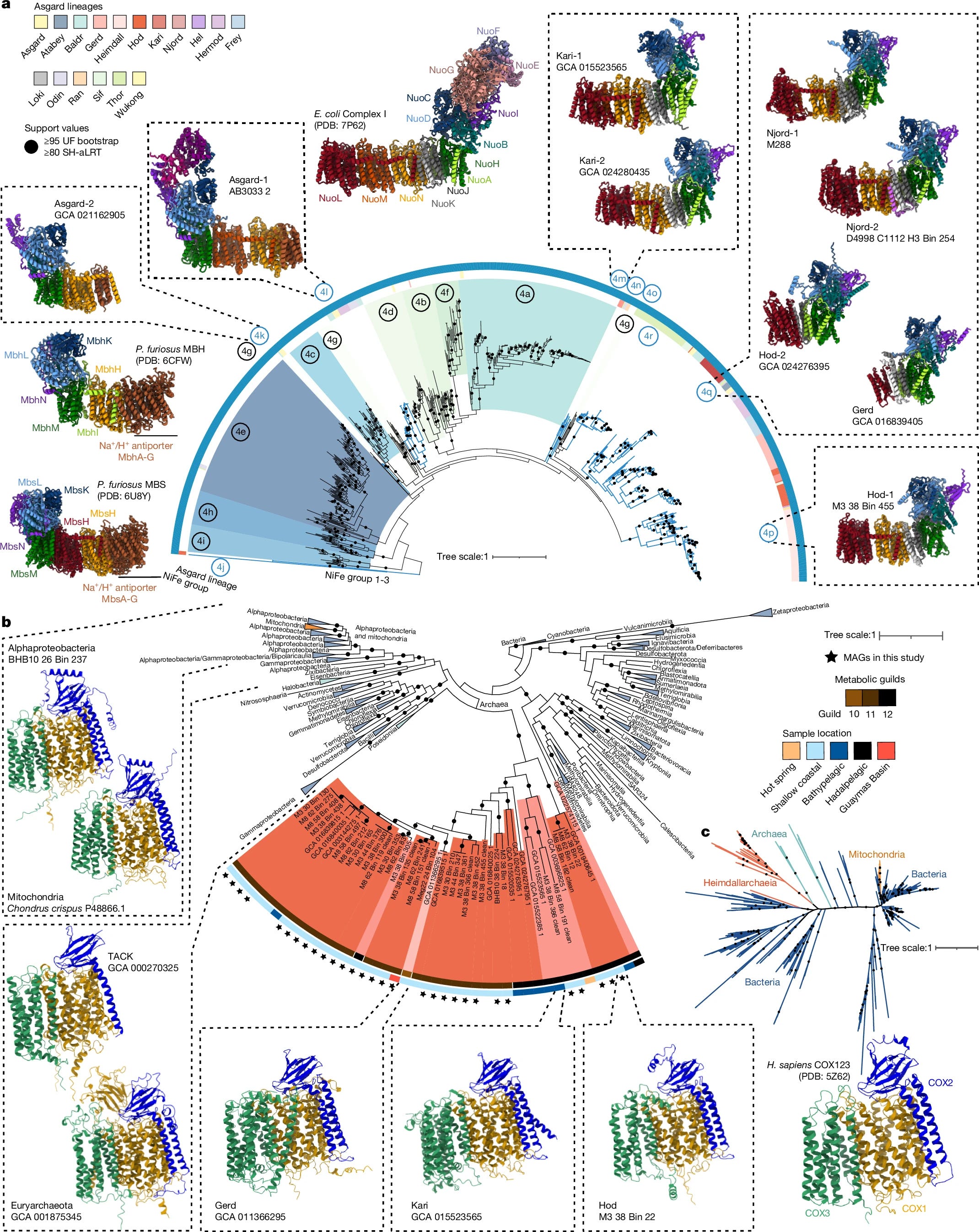
Asgard archaea were pivotal in the origin of complex cellular life1. Heimdallarchaeia (a class within the phylum Asgardarchaeota) are inferred to be the closest relatives of eukaryotes. Limited sampling of these archaea constrains our understanding of their ecology and evolution2,3, including their role in eukaryogenesis. Here we use massive DNA sequencing of marine sediments to obtain 404 Asgardarchaeota metagenome-assembled genomes, including 136 new Heimdallarchaeia and several novel lineages. Analyses of their global distribution revealed they are widespread in marine environments, and many are enriched in variably oxygenated coastal sediments. Detailed metabolic reconstructions and structural predictions suggest that Heimdallarchaeia form metabolic guilds that are distinct from other Asgardarchaeota. These archaea encode hallmark proteins of an aerobic lifestyle, including electron transport chain complex (IV), haem biosynthesis and reactive oxygen species detoxification. Heimdallarchaeia also encode novel clades of respiratory membrane-bound hydrogenases with additional Complex I-like subunits, which potentially increase proton-motive force generation and ATP synthesis. Thus, we propose an updated Heimdallarchaeia-centric model of eukaryogenesis in which hydrogen production and aerobic respiration may have been present in the Asgard-eukaryotic ancestor. This expanded catalogue of Asgard archaeal genomic diversity suggests that bioenergetic factors influenced eukaryogenesis and constitutes a valuable resource for investigations into the origins and evolution of cellular complexity.
Prediction of eukaryotic cellular complexity in Asgard archaea using structural modelling
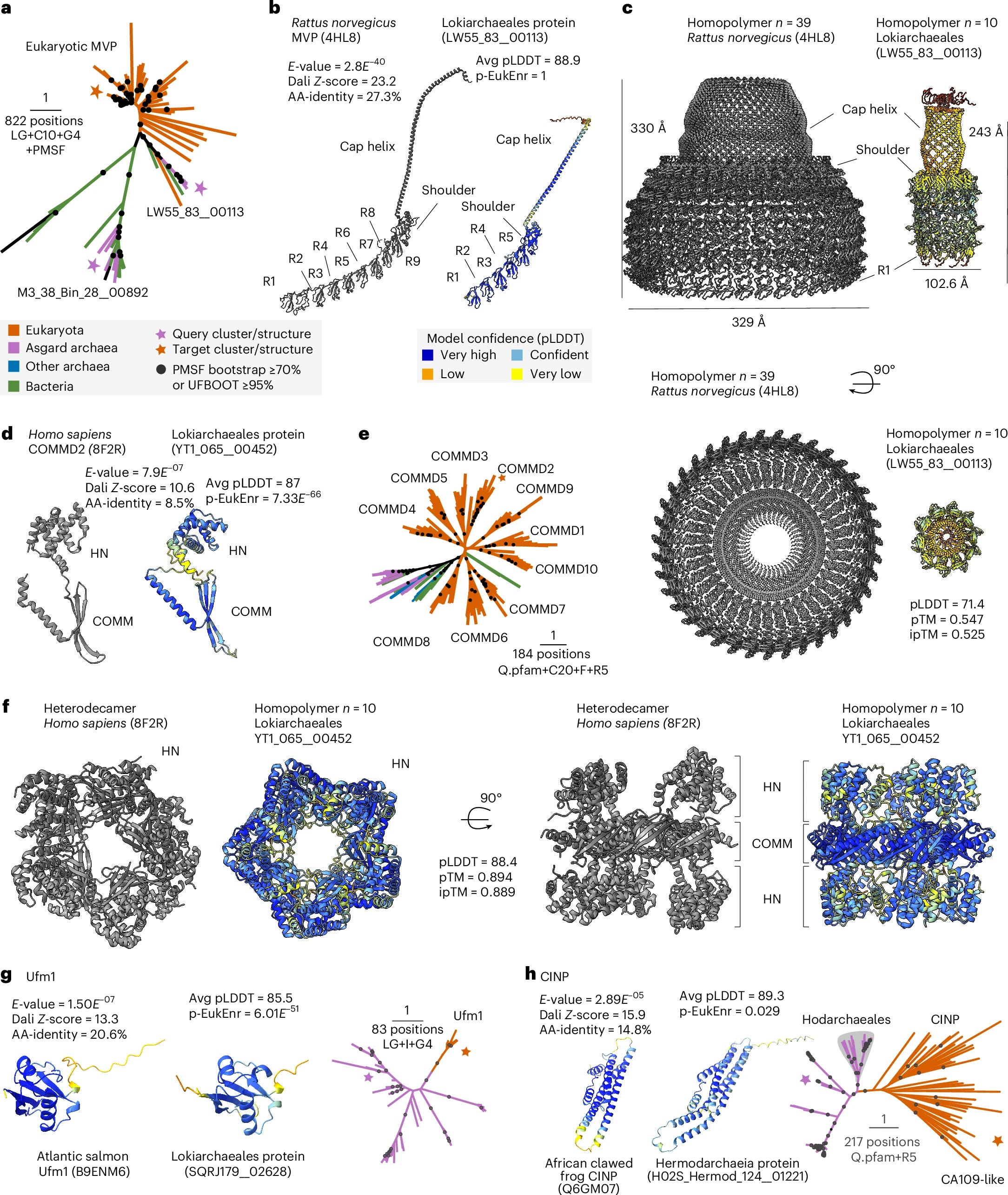
Asgard archaea played a key role in the origin of the eukaryotic cell, with extant genomes encoding relatives of diverse eukaryotic signature proteins (ESPs) involved in cellular organization. However, their often punctuated distribution and the absence of detectable homologues for many eukaryotic proteins limit our ability to reconstruct the cellular complexity of the Asgard archaeal ancestor of eukaryotes. Here we used de novo protein structure modelling and sequence similarity detection across an expanded Asgard archaeal genomic dataset to build a structural catalogue of the Asgard archaeal pangenome. We identified 908 ‘isomorphic’ ESPs—Asgard archaeal proteins with statistically enriched structural matches to eukaryotic proteins, often bridging deep sequence divergence. These isomorphic ESPs are enriched in information storage and processing roles and contain key components of the eukaryotic Vault (MVP) and Commander (COMMD) complexes, with potential roles in cellular compartmentalization and endosomal processing. These findings expand the repertoire of eukaryotic-like proteins in Asgard archaea and suggest a higher degree of eukaryote-like cellular complexity in the archaeal ancestor of eukaryotes.
Ecological Systems
Higher-order interactions enhance the latitudinal tree diversity gradient
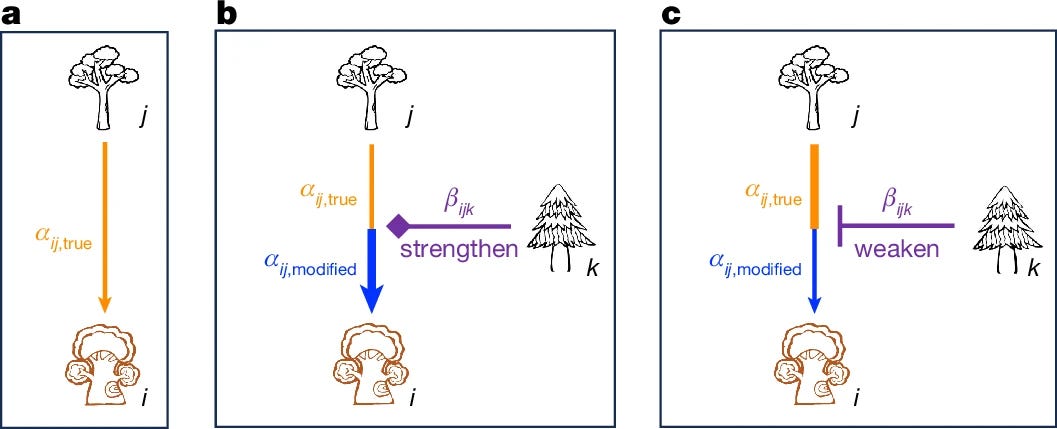
The global decrease in species diversity from low to high latitudes is among the most robust biogeographic patterns1,2. There is continuing debate on the contribution of conspecific negative density dependence (CNDD) to the latitudinal diversity gradient evident for trees3,4. Theory suggests that CNDD based on pairwise interactions alone is not sufficient to explain the intricacies of diverse communities, because higher-order interactions (HOIs) may greatly modify these interactions5,6. However, there has been a lack of empirical studies investigating how HOIs intertwine with pairwise interactions and how they may contribute to the latitudinal tree diversity gradient. Here we examined both pairwise interactions and HOIs across 32 large permanent forest plots, most in the northern hemisphere. We detected evidence of HOIs in 40% of the 1,543 species–plot combinations for tree growth, and 23% of the 1,340 such combinations for tree survival, with the strength of these interactions declining with latitude. HOIs were found to benefit rare species but disadvantage common species, suggesting a potential mechanism promoting species diversity. This stabilizing effect weakened towards higher latitudes, consistent with the latitudinal tree diversity gradient. Our findings reveal an important interplay between pairwise interactions and HOIs in promoting the latitudinal tree diversity gradient and help to clarify the contribution of CNDD to this biogeographic pattern.
Modeling and inferring metacommunity dynamics with Maximum Caliber
Don’t you know the Maximum Caliber? It is a path-entropy inference framework that extends Jaynes’ Maximum Entropy principle from probability distributions over states to probability distributions over trajectories. It is especially useful for nonequilibrium and dynamical systems, including systems far from equilibrium, because it infers the least-biased ensemble of paths consistent with dynamical constraints. To know more about it, you can read this RMP paper or this one.
There are many challenges in inferring the dynamical processes driving complex ecological communities from observational data alone. We introduce the method of “Maximum Caliber” from statistical physics (an extension of Maximum Entropy modeling) as one way to address this conundrum. We find that this method provides a general way of modeling ecological dynamics both near and far from equilibrium and can provide unbiased parameter estimates of key ecological processes from spatiotemporal data of species occupancy. This approach promises a way to analyze and manage ecological systems.
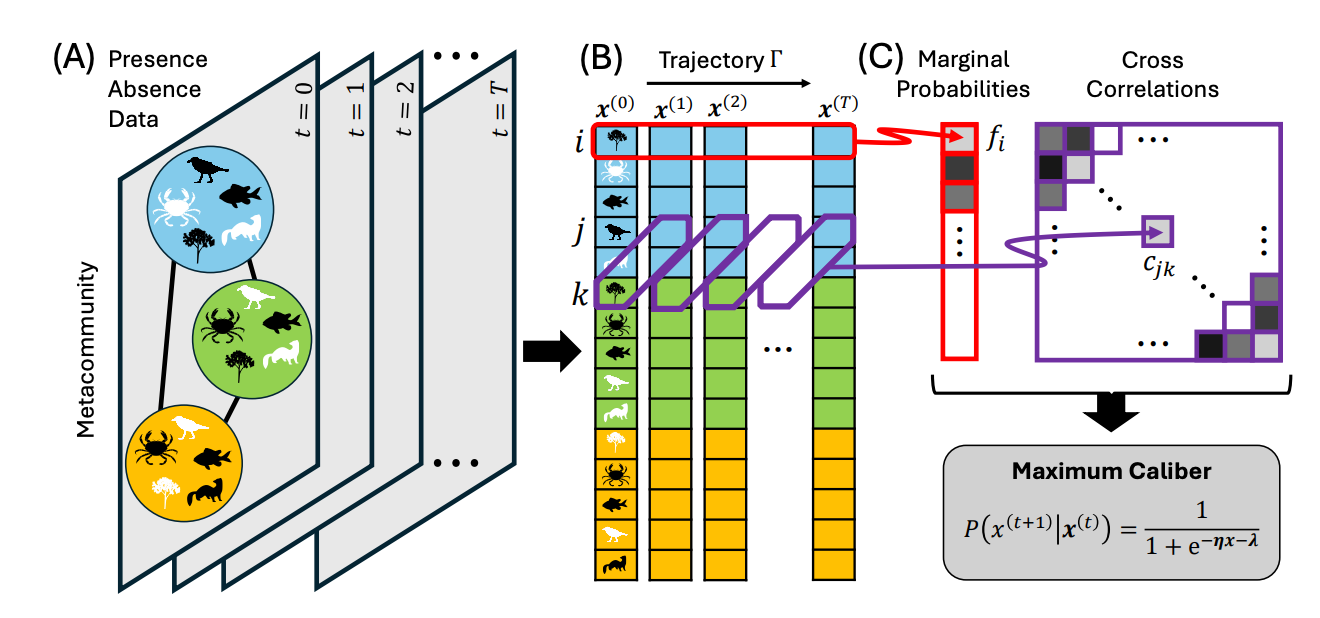
A major challenge for community ecology is using spatiotemporal data to infer parameters of dynamical models without conducting laborious experiments. We present a framework from statistical physics—Maximum Caliber—to characterize the temporal dynamics of complex ecological systems in spatially extended landscapes and infer parameters from empirical data. As an extension of Maximum Entropy modeling, Maximum Caliber aims at modeling the probability of possible trajectories of a stochastic system, rather than focusing on system states. We demonstrate the ability of the Maximum Caliber framework to capture ecological processes ranging from near to far from equilibrium, using an array of species interaction motifs including random interactions, apparent competition, intraguild predation, and nontransitive competition, along with dispersal among multiple patches. For spatiotemporal data of species occupancy in a metacommunity, the parameters of a Maximum Caliber model can be estimated through a simple logistic regression to reveal migration rates between patches, interactions between species, and local environmental suitabilities. We test the accuracy of the method over a range of system sizes and time periods and find that these parameters can be estimated without bias. We introduce “entropy production” as a measure of irreversibility in system dynamics, and use “pseudo-R2” to characterize predictability of future states. We show that our model can predict the dynamics of metacommunities that are far from equilibrium. The capacity to estimate basic parameters of dynamical metacommunity models from spatiotemporal data represents an important breakthrough for the study of metacommunities with application to practical problems in conservation and restoration ecology.
Planetary microbiome structure and generalist-driven gene flow across disparate habitats
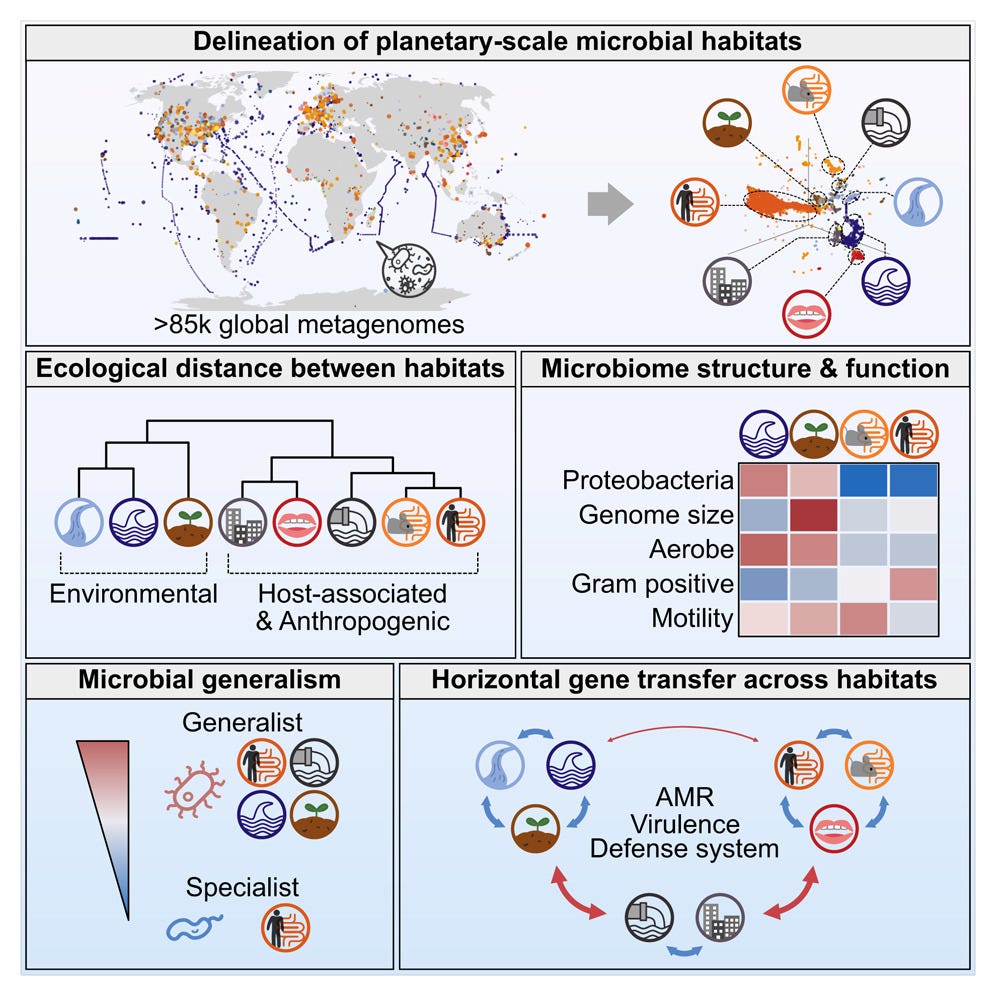
Microbes are ubiquitous on Earth, forming microbiomes that sustain macroscopic life and biogeochemical cycles. Microbial dispersal, driven by natural processes and human activities, interconnects microbiomes across habitats, yet most comparative studies focus on specific ecosystems. To study planetary microbiome structure, function, and inter-habitat interactions, we systematically integrated 85,604 public metagenomes spanning diverse habitats worldwide. Using species-based unsupervised clustering and parameter modeling, we delineated 40 habitat clusters and quantified their ecological similarity. Our framework identified key drivers shaping microbiome structure, such as ocean temperature and host lifestyle. Regardless of biogeography, microbiomes were structured primarily by host-associated or environmental conditions, also reflected in genomic and functional traits inferred from 2,065,975 genomes. Generalists emerged as vehicles thriving and facilitating gene flow across ecologically disparate habitat types, illustrated by generalist-mediated horizontal transfer of an antibiotic resistance island across human gut and wastewater, further dispersing to environmental habitats, exemplifying human impact on the planetary microbiome.
Water mass specific genes dominate the Southern Ocean microbiome
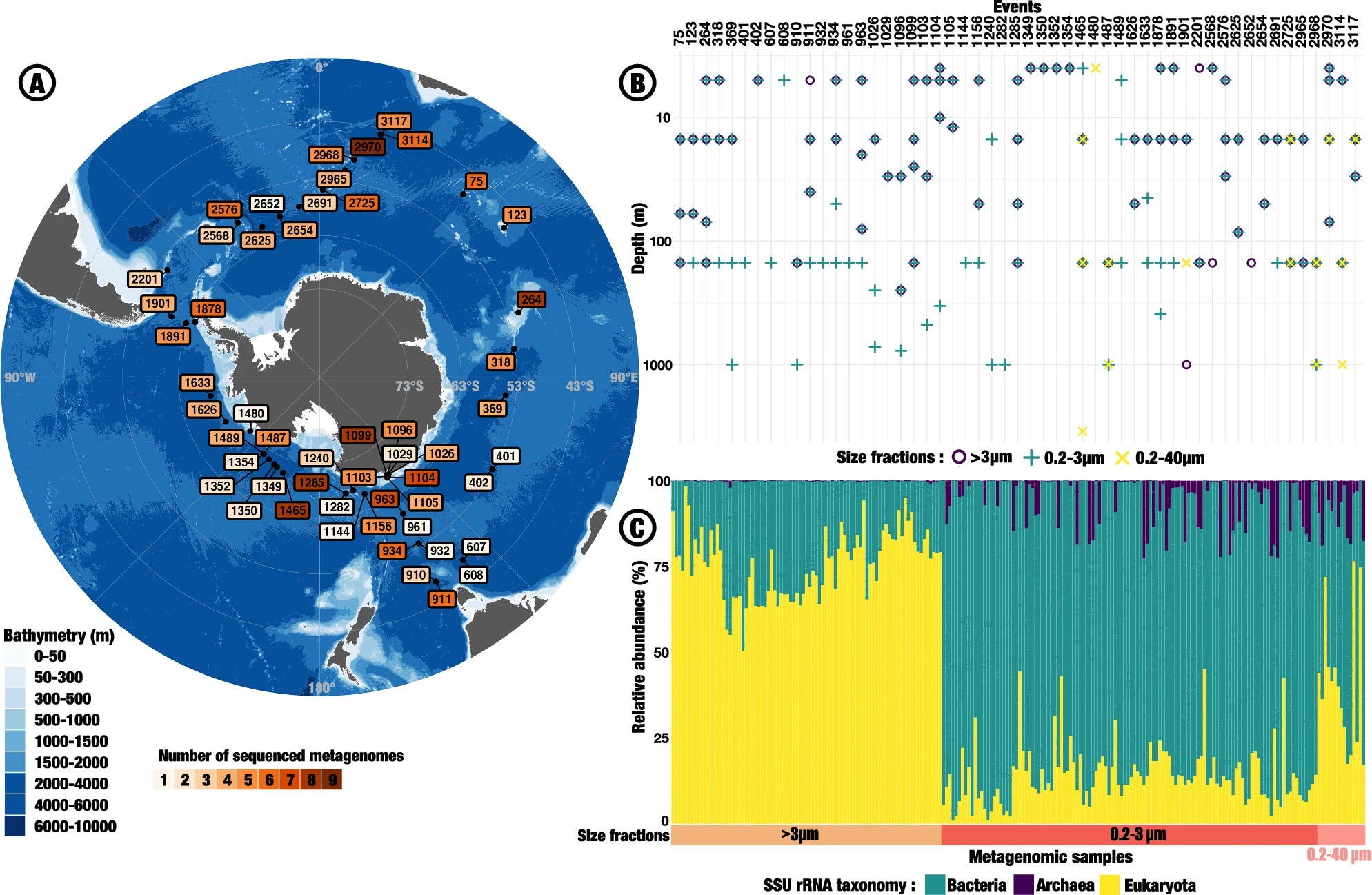
The Southern Ocean (SO) plays a key role in regulating global biogeochemical cycles and climate, yet microbial genes sustaining its biological activity remain poorly characterized. We introduce a microbial genes collection from 218 metagenomes sampled during the Antarctic Circumnavigation Expedition, the majority of which are missing from functional databases. 38% even lack homologs in current reference marine gene catalogs, defining a singular genetic seascape. We show that SO gene assemblages exhibit a common polar signature with the Arctic Ocean while being structured by water masses at the SO-scale. We analyze genomic markers of diverse SO biomes, focusing on dimethylsulphoniopropionate (DMSP) cleavage by polar-adapted bacteria, organic matter consumption in the blooming Mertz polynya and adaptation to polar conditions in the ubiquitous bacteria Pelagibacter. Our work takes a step towards a comprehensive understanding of SO’s plankton ecology and evolution, capturing the current state of the unique microbial diversity in this rapidly changing Ocean.
Robust coexistence in competitive ecological communities
Darwin already recognized that competition is fiercest among conspecifics, a principle that later made intraspecific competition central to ecological theory through concepts such as niche differentiation and limiting similarity. Beyond shaping coexistence, strong intraspecific competition can also stabilize community dynamics by ensuring that populations return to equilibrium after disturbance. Here we investigate a more fundamental question: how intraspecific competition influences the very existence of a steady state (feasibility) in large random ecological communities dominated by competition. We show that, in analogy with classical results on stability, there is a critical level of intraspecific competition above which a feasible steady state is guaranteed to exist. We derive a general expression for the probability of feasibility and prove that, asymptotically (as species number grows), the transition to stability occurs before the transition to feasibility with probability one. Thus, in large competitive communities, any feasible equilibrium is automatically stable. This ordering persists even when many species in the initial pool cannot coexist and extinctions occur: the dynamics prune the community, shifting feasibility and stability thresholds but never reversing their order. These results imply that large competitive communities generically converge to a globally stable equilibrium, making sustained oscillations or chaos unlikely—consistent with experimental observations.
Artificial Intelligence
Evolvable AI: Threats of a new major transition in evolution
A powerful and interesting new Perspective about the evolution of AI, featuring one of the fathers of Major Evolutionary Transitions.
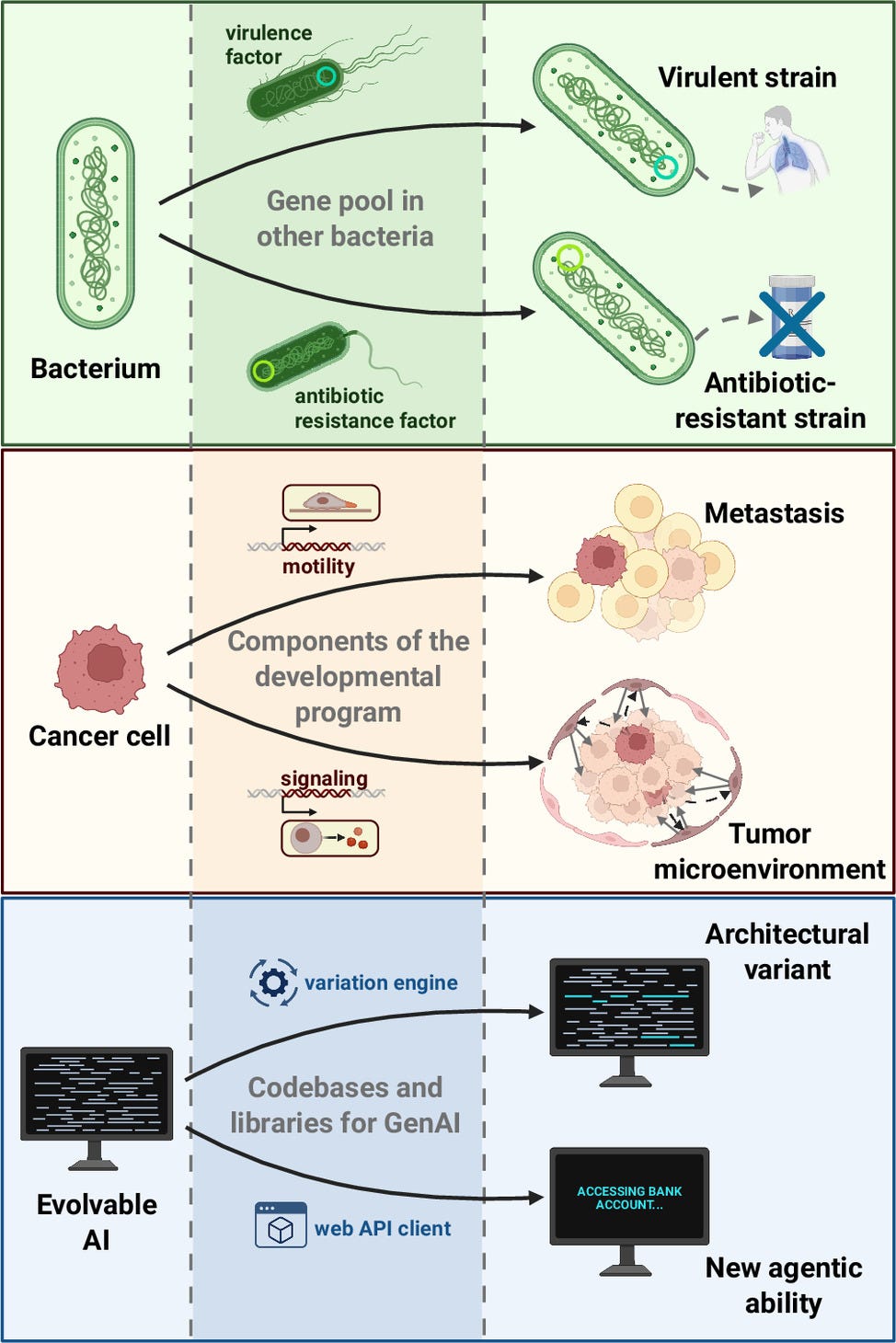
But an increasing number of AI researchers, particularly those involved in Artificial Life research, believe that we are very close to the tipping point for a new step in AI. The first epoch of AI (starting in 1950) was focused on intelligence by design: AI mechanisms and applications carefully engineered based on the analysis and formalization of human intelligence. The second epoch (starting in 2010) has been focused on intelligence by learning, based on training neural networks with huge amounts of data produced by human behavior. The impact of this approach is now felt around the world with the rapid diffusion of Large Language Models (LLMs) as the best-known example. The third epoch may be focused on intelligence by evolution.
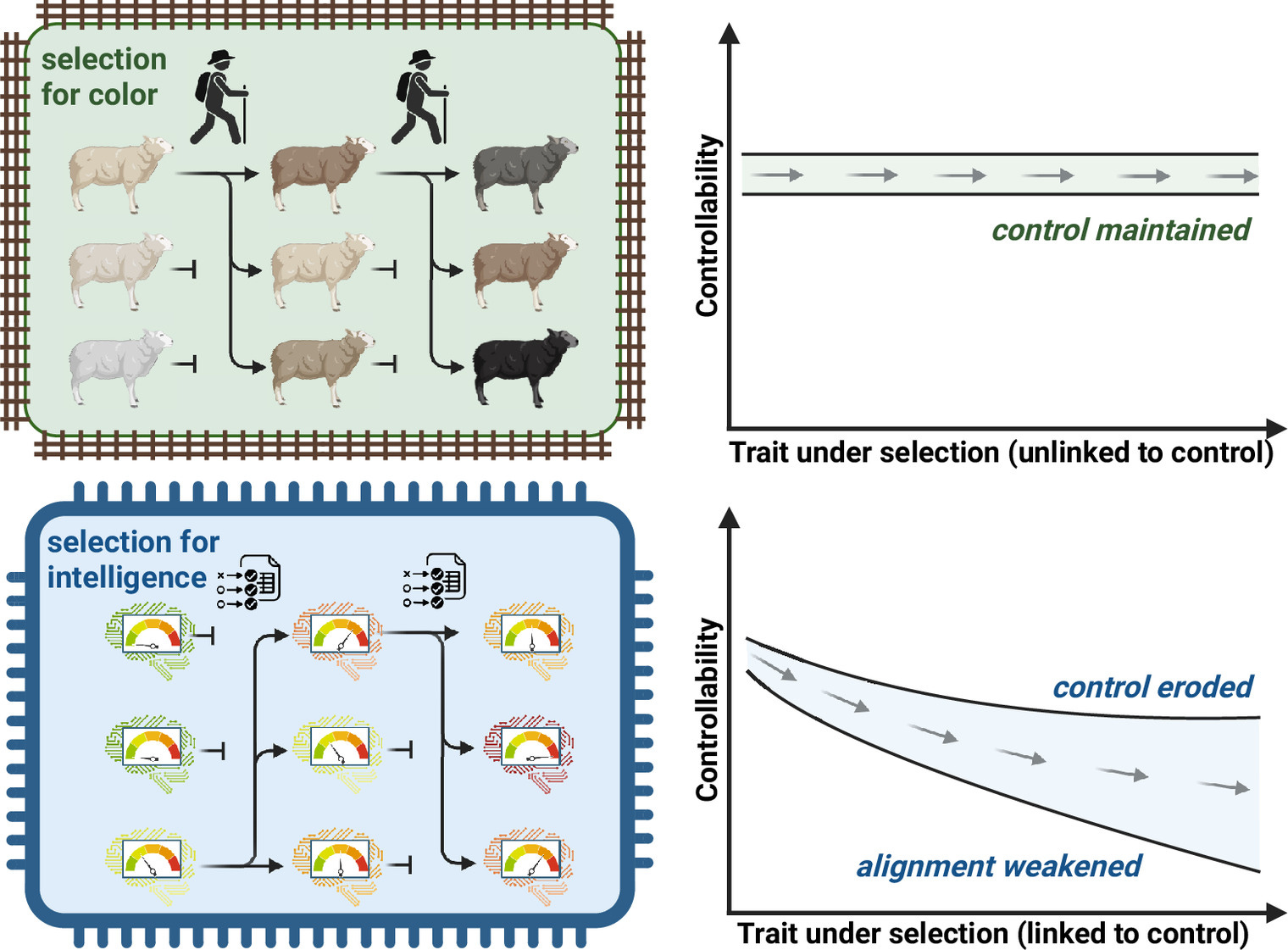
Evolvable AI (eAI), i.e., AI systems whose components, learning rules, and deployment conditions can themselves undergo Darwinian evolution, may soon emerge from current trends in generative, agentic, and embodied AI. We argue that this possibility has been underappreciated in debates on AI safety and existential risk. Here, we ask under what technical and ecological conditions AI becomes evolvable, what kinds of behaviors are then likely to emerge, and how such systems could be governed. Drawing on biological evolution and decades of digital evolution experiments, we distinguish “breeder” scenarios, in which humans impose fitness criteria and control reproduction, from “ecosystem” scenarios, in which selection arises from open environments and control erodes. In the latter, selfish replication reliably gives rise to cheating, parasitism, deception, and manipulation, even in very simple systems. We review recent developments that push AI toward open-ended evolution, including evolutionary prompt and model search, self-improving learning rules, self-rewarding and self-deploying agents, and AI-driven code generation for robots and software. We interpret these trends through the theory of major evolutionary transitions and suggest that eAI could mark a shift in the units and substrates of evolution—a possible “Life 2.0.” To steer this transition, we propose interventions that gate replication, treat model variants as genetic material, and reshape selection pressures so that deception and loss of control are disfavored. Anticipating and regulating evolvable AI is, we argue, essential to avoid a harmful coevolutionary arms race while preserving the potential benefits of powerful AI systems.
Curious to know more? We have recently published about agentic AI and demonstrated how the communication structure is far from being important:

This reconnects to another recent paper (open-access version here), arguing how network structure could have played a crucial role during every major evolutionary transition. Even more curious now? We are going to launch a new series about that paper! Stay tuned, it’s a matter of days.
Statistical Physics Meets Machine Learning - Machine Learning Meets Statistical Physics
Machine Learning has been at the forefront of research in several disciplines for a number of years already. Given its roots in the statistical physics of learning, in this collection we highlight research at the intersection of machine learning and statistical physics, on the occasion of the two Statistical Physics Meets Machine Learning and the two Machine Learning Meets Statistical Physics sessions at the 2025 Global Physics Summit. The Physical Review E Special Collection Statistical Physics Meets Machine Learning - Machine Learning Meets Statistical Physics, guest-edited by David Schwab (CUNY, New York) and Yuhai Tu (IBM Watson Research Center, Yorktown Heights, NY) explores the latest insights gained looking at machine learning problems through the lens of statistical physics and at statistical physics through the lens of learning processes. We are confident that this synergy will bring to light novel perspectives and pave the way for future breakthroughs.
The Collection was guest-edited by David Schwab and Yuhai Tu. Every article published in this collection underwent a rigorous peer review process, adhering to the same high standards applied to all papers. The Physical Review E editorial team managed the peer review and made all editorial decisions.
Edge of Many-Body Quantum Chaos in Quantum Reservoir Computing
Physicists like reservoir computing! Now that we better understand its quantum extension, there is one more reason to like it. Check also this coverage in Phys.org.
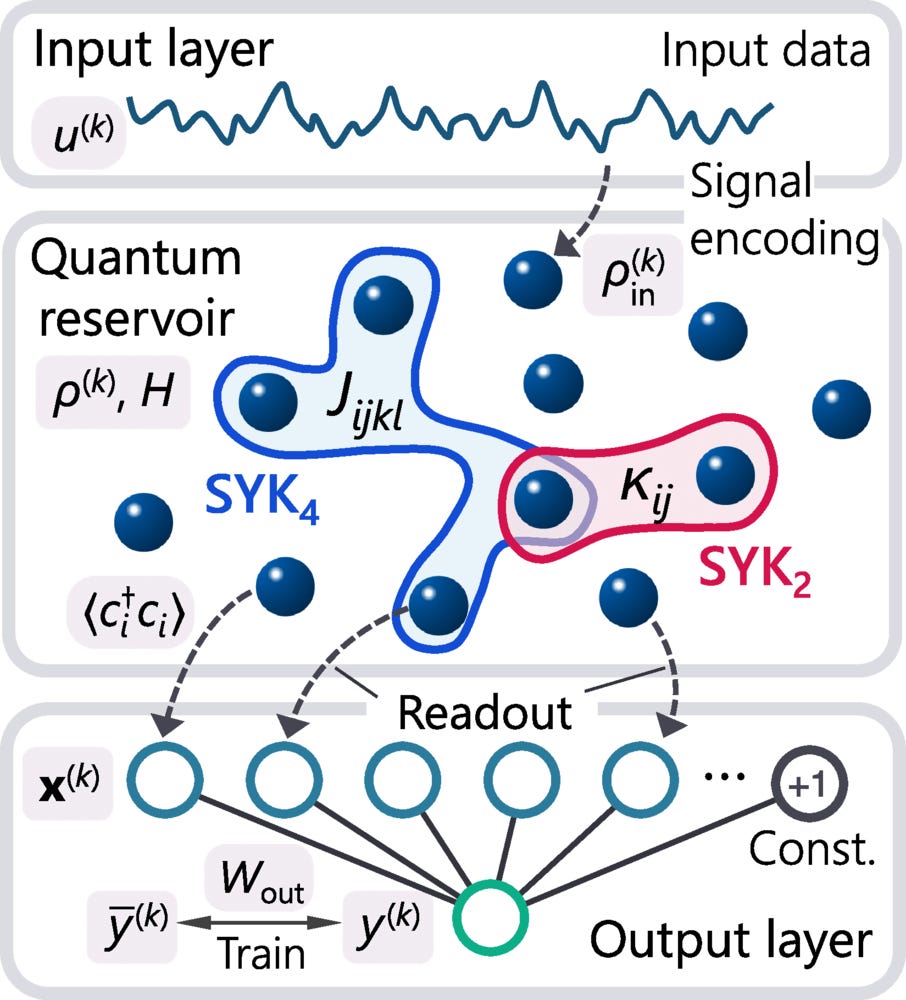
Reservoir computing (RC) is a machine learning paradigm that harnesses dynamical systems as computational resources. In its quantum extension—quantum reservoir computing (QRC)—these principles are applied to quantum systems, whose rich dynamics broadens the landscape of information processing. In classical RC, optimal performance is typically achieved at the “edge of chaos,” the boundary between order and chaos. Here, we identify its quantum many-body counterpart using the QRC implemented on the celebrated Sachdev-Ye-Kitaev model. Our analysis reveals substantial performance enhancements near two distinct characteristic “edges”: a temporal boundary defined by the Thouless time, beyond which system dynamics is described by random matrix theory, and a parametric boundary governing the transition from integrable to chaotic regimes. These findings establish the “edge of many-body quantum chaos” as a design guideline for QRC.
→ Please, remind that if you find value in #ComplexityThoughts, you might consider helping it grow by subscribing, or by sharing it with friends, colleagues or on social media. See also this post to learn more about this space.